As always, you can directly jump for checking TL;DR first.
1. What’s inside the *.tissue.gef?
Let’s first explore the resulted <SN>.tissue.gef in Python:
import h5py
import numpy as np
def explore_gef(h5file):
def visitor(name, obj):
if isinstance(obj, h5py.Dataset):
print(f"{name}: shape={obj.shape}, dtype={obj.dtype}")
with h5py.File(h5file, 'r') as f:
f.visititems(visitor)
gef_file = "./<SN>.tissue.gef"
with h5py.File('./<SN>.tissue.gef', 'r') as f:
def visit(name):
print(name)
f.visit(visit)Output:
contour
contour/tissueContour
geneExp
geneExp/bin1
geneExp/bin1/exon
geneExp/bin1/expression
geneExp/bin1/gene
geneExp/bin10
geneExp/bin10/exon
geneExp/bin10/expression
geneExp/bin10/gene
geneExp/bin100
geneExp/bin100/exon
geneExp/bin100/expression
geneExp/bin100/gene
geneExp/bin150
geneExp/bin150/exon
geneExp/bin150/expression
geneExp/bin150/gene
geneExp/bin20
geneExp/bin20/exon
geneExp/bin20/expression
geneExp/bin20/gene
geneExp/bin200
geneExp/bin200/exon
geneExp/bin200/expression
geneExp/bin200/gene
geneExp/bin5
geneExp/bin5/exon
geneExp/bin5/expression
geneExp/bin5/gene
geneExp/bin50
geneExp/bin50/exon
geneExp/bin50/expression
geneExp/bin50/gene
stat
stat/gene
wholeExp
wholeExp/bin1
wholeExp/bin10
wholeExp/bin100
wholeExp/bin150
wholeExp/bin20
wholeExp/bin200
wholeExp/bin5
wholeExp/bin50
wholeExpExon
wholeExpExon/bin1
wholeExpExon/bin10
wholeExpExon/bin100
wholeExpExon/bin150
wholeExpExon/bin20
wholeExpExon/bin200
wholeExpExon/bin5
wholeExpExon/bin50Let’s check where <SN>.tissue.gef file stores the XY coordinates continuous in Python:
explore_gef(gef_file)Output:
contour/tissueContour: shape=(), dtype=object
geneExp/bin1/exon: shape=(116772117,), dtype=uint8
geneExp/bin1/expression: shape=(116772117,), dtype=[('x', '<i4'), ('y', '<i4'), ('count', 'u1')]
geneExp/bin1/gene: shape=(57584,), dtype=[('geneID', 'S64'), ('geneName', 'S64'), ('offset', '<u4'), ('count', '<u4')]
geneExp/bin10/exon: shape=(93972549,), dtype=uint16
geneExp/bin10/expression: shape=(93972549,), dtype=[('x', '<i4'), ('y', '<i4'), ('count', '<u2')]
geneExp/bin10/gene: shape=(57584,), dtype=[('geneID', 'S64'), ('geneName', 'S64'), ('offset', '<u4'), ('count', '<u4')]
geneExp/bin100/exon: shape=(32675685,), dtype=uint16
geneExp/bin100/expression: shape=(32675685,), dtype=[('x', '<i4'), ('y', '<i4'), ('count', '<u2')]
geneExp/bin100/gene: shape=(57584,), dtype=[('geneID', 'S64'), ('geneName', 'S64'), ('offset', '<u4'), ('count', '<u4')]
geneExp/bin150/exon: shape=(22060836,), dtype=uint16
geneExp/bin150/expression: shape=(22060836,), dtype=[('x', '<i4'), ('y', '<i4'), ('count', '<u2')]
geneExp/bin150/gene: shape=(57584,), dtype=[('geneID', 'S64'), ('geneName', 'S64'), ('offset', '<u4'), ('count', '<u4')]
geneExp/bin20/exon: shape=(78271410,), dtype=uint16
geneExp/bin20/expression: shape=(78271410,), dtype=[('x', '<i4'), ('y', '<i4'), ('count', '<u2')]
geneExp/bin20/gene: shape=(57584,), dtype=[('geneID', 'S64'), ('geneName', 'S64'), ('offset', '<u4'), ('count', '<u4')]
geneExp/bin200/exon: shape=(15879595,), dtype=uint16
geneExp/bin200/expression: shape=(15879595,), dtype=[('x', '<i4'), ('y', '<i4'), ('count', '<u2')]
geneExp/bin200/gene: shape=(57584,), dtype=[('geneID', 'S64'), ('geneName', 'S64'), ('offset', '<u4'), ('count', '<u4')]
geneExp/bin5/exon: shape=(103779528,), dtype=uint8
geneExp/bin5/expression: shape=(103779528,), dtype=[('x', '<i4'), ('y', '<i4'), ('count', 'u1')]
geneExp/bin5/gene: shape=(57584,), dtype=[('geneID', 'S64'), ('geneName', 'S64'), ('offset', '<u4'), ('count', '<u4')]
geneExp/bin50/exon: shape=(52649679,), dtype=uint16
geneExp/bin50/expression: shape=(52649679,), dtype=[('x', '<i4'), ('y', '<i4'), ('count', '<u2')]
geneExp/bin50/gene: shape=(57584,), dtype=[('geneID', 'S64'), ('geneName', 'S64'), ('offset', '<u4'), ('count', '<u4')]
stat/gene: shape=(57584,), dtype=[('geneID', 'S64'), ('geneName', 'S64'), ('MIDcount', '<u4'), ('E10', '<f4')]
wholeExp/bin1: shape=(26460, 26460), dtype=[('MIDcount', 'u1'), ('genecount', '<u2')]
wholeExp/bin10: shape=(2646, 2646), dtype=[('MIDcount', '<u2'), ('genecount', '<u2')]
wholeExp/bin100: shape=(265, 265), dtype=[('MIDcount', '<u2'), ('genecount', '<u2')]
wholeExp/bin150: shape=(177, 177), dtype=[('MIDcount', '<u4'), ('genecount', '<u2')]
wholeExp/bin20: shape=(1323, 1323), dtype=[('MIDcount', '<u2'), ('genecount', '<u2')]
wholeExp/bin200: shape=(133, 133), dtype=[('MIDcount', '<u4'), ('genecount', '<u2')]
wholeExp/bin5: shape=(5292, 5292), dtype=[('MIDcount', '<u2'), ('genecount', '<u2')]
wholeExp/bin50: shape=(530, 530), dtype=[('MIDcount', '<u2'), ('genecount', '<u2')]
wholeExpExon/bin1: shape=(26460, 26460), dtype=uint8
wholeExpExon/bin10: shape=(2646, 2646), dtype=uint16
wholeExpExon/bin100: shape=(265, 265), dtype=uint16
wholeExpExon/bin150: shape=(177, 177), dtype=uint32
wholeExpExon/bin20: shape=(1323, 1323), dtype=uint16
wholeExpExon/bin200: shape=(133, 133), dtype=uint32
wholeExpExon/bin5: shape=(5292, 5292), dtype=uint16
wholeExpExon/bin50: shape=(530, 530), dtype=uint16Now, we know that the <SN>.tissue.gef file stores the XY coordinates in geneExp/bin1/expression on the smallest level. Therefore, we will save them out into tissue_xy_coords.txt for later use by using Python:
with h5py.File(gef_file, 'r') as f:
expression = f['geneExp/bin1/expression']
x_coords = expression['x']
y_coords = expression['y']
xy_set = set(zip(x_coords, y_coords))
# Save to tab-separated list
with open('tissue_xy_coords.txt', 'w') as out:
for x, y in sorted(xy_set):
out.write(f"{x}\t{y}\n")Check the output tissue_xy_coords.txt:
ubuntu4@ubuntu4:~$ head tissue_xy_coords.txt
5713 26441
5787 26358
5788 26318
5788 26319
5788 26321
5788 26329
5789 26316
5789 26327
5790 26305
5790 263102. The —clean-reads-fastq option in saw software
Actually, with the —clean-reads-fastq option in saw software, we can get the read ID along with the XY coordinates and the biological sequence originally from Read2, as shown below:
ubuntu@ubuntu:~$ head <ID>_<LaneNo.>_<SampleNo.>_1.clean_reads.fq
@E150018299L1C004R03402098317|Cx:i:19892|Cy:i:17727 E8BBE63524CF BEAB3
GCCAATAGTATAGTGTGGTGTGCTTTTACGTGATGGCGAGTGGGCAGCGGGCGGTGGGCTGTACACAGCCGTCTGTCCTTTGAATCTCAATCTGCCTGCG
+
9A>CCCCACCCC>CACB?CCC>ACCCCCA>C@<AB8<ABB?>B;6BC7?/8=B>ACA=>CC@BAB<BC=CCCACABB<CCCC1CCAC8C/A;CCCCC@CA
@E150018299L1C004R03402098337|Cx:i:13026|Cy:i:14469 1C20554DC029 A5B9F
GCGTGGCACTCAGGCGGGGCCCTGGGAGCGCTGCGGGCACGGGGTGGCCGGCAGGACGCGGGCTGGATGGCTCTGGCCGCGCCAGGAGGAGGCCGACCTG
+
1B1AC>ABC<CCCBB8<C0>>1C;A95C:>9>@869<2C?@C;C1C?A=ACA@CC6BC=CA>6CAA:BCC?BCCCA<1@CCA:@CA1<A)CC93-CCCCC
@E150018299L1C004R03402098484|Cx:i:17752|Cy:i:16736 8E7513DF377D FD27F
GCCGGGGGCATTCGTATTGCGCCGCTAGAGGTGAAATTCTTGGACCGGCGCAAGACGGACCAGAGCGAAAGCATTTGCCAAGAATGTTTTCATTAATCAATherefore, we can use the tissue_xy_coords.txt to filter the *_1.clean_reads.fq files directly into a merged R2.tissue.fq.gz file by using the filter_cleanR1_by_XY_parallel.sh below:
#!/bin/bash
# ===== CONFIG =====
XY_LIST="tissue_xy_coords.txt"
OUTFILE="R2.tissue.fq.gz"
TMPDIR="tmp_cleanR1"
mkdir -p "$TMPDIR"
echo "📄 Preparing whitelist..."
# Extract and sort unique XY coordinates (Cx, Cy)
cut -f1,2 "$XY_LIST" | sort | uniq > whitelist_xy.txt
# Export for GNU parallel
export TMPDIR
export WL="$(realpath whitelist_xy.txt)" # Use full path to avoid parallel context issues
# ===== FUNCTION: PROCESS EACH FILE =====
process_cleanR1() {
fq="$1"
base=$(basename "$fq" .clean_reads.fq)
tmpout="$TMPDIR/${base}.tissue.fq"
gawk -v WL="$WL" -v OUT="$tmpout" '
BEGIN {
while ((getline line < WL) > 0) {
gsub(/\r/, "", line) # Remove Windows-style line endings
valid_xy[line] = 1
}
}
{
if (substr($0,1,1) == "@") {
id = $0
getline seq
getline plus
getline qual
if (match(id, /\|Cx:i:([0-9]+)\|Cy:i:([0-9]+)/, m)) {
xy = m[1] "\t" m[2]
if (xy in valid_xy) {
print id >> OUT
print seq >> OUT
print plus >> OUT
print qual >> OUT
}
}
}
}
' "$fq"
}
export -f process_cleanR1
# ===== PARALLEL PROCESSING =====
echo "🚀 Filtering reads based on XY coordinates..."
ls *_1.clean_reads.fq | parallel --bar -j "$(nproc)" process_cleanR1 {}
# ===== MERGE RESULTS =====
echo "🧬 Merging into $OUTFILE..."
if ls $TMPDIR/*.tissue.fq 1> /dev/null 2>&1; then
cat $TMPDIR/*.tissue.fq | gzip > "$OUTFILE"
echo "✅ Done. Output written to $OUTFILE"
else
echo "⚠️ No matching reads found. Output file was not created."
fi
Output:
ubuntu@ubuntu:~$ zcat R2.tissue.fq.gz|head -n 12
@E150018299L1C004R03402098317|Cx:i:19892|Cy:i:17727 E8BBE63524CF BEAB3
GCCAATAGTATAGTGTGGTGTGCTTTTACGTGATGGCGAGTGGGCAGCGGGCGGTGGGCTGTACACAGCCGTCTGTCCTTTGAATCTCAATCTGCCTGCG
+
9A>CCCCACCCC>CACB?CCC>ACCCCCA>C@<AB8<ABB?>B;6BC7?/8=B>ACA=>CC@BAB<BC=CCCACABB<CCCC1CCAC8C/A;CCCCC@CA
@E150018299L1C004R03402098337|Cx:i:13026|Cy:i:14469 1C20554DC029 A5B9F
GCGTGGCACTCAGGCGGGGCCCTGGGAGCGCTGCGGGCACGGGGTGGCCGGCAGGACGCGGGCTGGATGGCTCTGGCCGCGCCAGGAGGAGGCCGACCTG
+
1B1AC>ABC<CCCBB8<C0>>1C;A95C:>9>@869<2C?@C;C1C?A=ACA@CC6BC=CA>6CAA:BCC?BCCCA<1@CCA:@CA1<A)CC93-CCCCC
@E150018299L1C004R03402098484|Cx:i:17752|Cy:i:16736 8E7513DF377D FD27F
GCCGGGGGCATTCGTATTGCGCCGCTAGAGGTGAAATTCTTGGACCGGCGCAAGACGGACCAGAGCGAAAGCATTTGCCAAGAATGTTTTCATTAATCAA
+
CCCB=CCCCCCCCCCDCCCCCCCCCDCCCCCC@CCCCCCCCC8BBCCCBCCCCBBCCCCCBCCBCCCCCCCCCCCCCBCCBCCCCCCCCCCCCCCBCCCC
To here, we got what we want.
3. If you need un-clean reads
According to SAW User Manual V8.1:
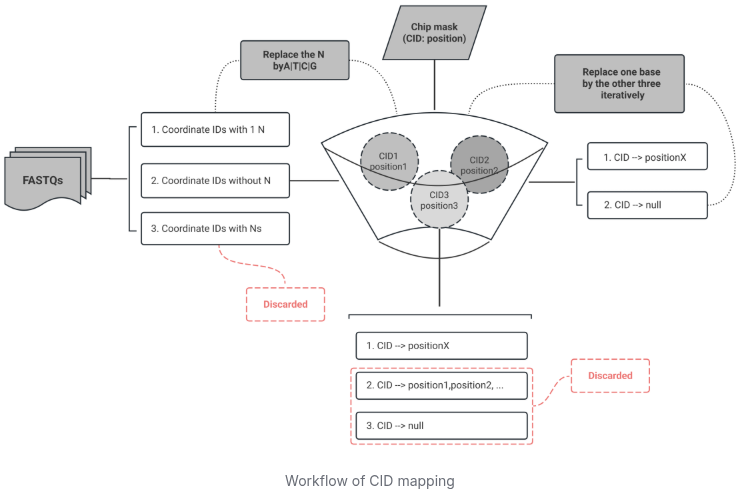
3.1. CID mapping
CID mapping requires FASTQs and a chip mask file, recording position information for sequencing reads.
Check the amount of Ns in Coordinate IDs:
- If there is 1 N base in CID, the N base will be replaced by
A/T/C/G. - CIDs without N bases will be directly poured into the match pool.
- CIDs with more than one N base will be discarded.
In the absense of an N base, if a CID does not match to any positions, each base of the CID will replaced by the other three types iterately until a successful match. After the mapping, only the unique CID match will be retained for subsequent steps.
3.2. RNA filtering
Before genome alignment, it is necessary to confirm that the reads entering the next step are cDNA sequences. Ideally, this part of the data only contains cDNA fragments, but there may be cases where the fragmented cDNA is too short or some non-cDNA fragments are detected.
Reads will be discarded if any of the following conditions are triggered:
- with a length of less than 30 after cutting out adapter sequences,
- mapped to DNB sequences,
- with a length of less than 30 after cutting out the poly-A sequence.
The above three are collectively known as “non-relevant short reads”.
3.3. MID filtering
MID sequences will be filtered out if they match any of the following:
- having more than one base with quality ⇐ Q10,
- having N bases >= 1.
In summary, the —clean-reads-fastq option lets the SAW software output the Clean Reads (before RNA alignment) in FASTQ format, which have undergone CID mapping, RNA filtering, and MID filtering.
Therefore, if you also need the reads before CID mapping, RNA filtering, and MID filtering (unclean reads), you can get if by following the below steps:
3.4. Extract Raw Reads by Barcodes
Let’s check the content of <SN>.barcodeToPos.h5 in the first by using Python:
3.4.1 What’s inside the <SN>.barcodeToPos.h5?
import h5py
import numpy as np
with h5py.File("<SN>.barcodeToPos.h5", "r") as f:
def show(name):
print(name)
f.visit(show)Output:
bpMatrix_1def visit_func(name, obj):
if isinstance(obj, h5py.Dataset):
print(f"{name}: shape={obj.shape}, dtype={obj.dtype}")
with h5py.File("<SN>.barcodeToPos.h5", "r") as f:
f.visititems(visit_func)Output:
bpMatrix_1: shape=(26462, 26462, 1), dtype=uint64with h5py.File("<SN>.barcodeToPos.h5", "r") as f:
bp_matrix = f["bpMatrix_1"]
preview = bp_matrix[:10, :10, 0] # First 10x10 values from the first "layer"
print(preview)Output:
[[ 836120372779751 26690364227217 310455682980154 810417757238641 757835266385246 468131266122076 730298897644317
114211586351941 905790851940934 144665928054079]
[ 592167366271184 315691865553282 539954256853234 637943645074939 638218522981883 935068209817518 943872959887278
145802161834220 384478059976677 90995135132138]
[ 473147532191787 916598384181374 373439451720958 911457121349196 946003516929503 701023829742140 0
1973895908933 482823435537541 882002041392058]
[ 75850009055013 497328043110101 157141292161565 912556632976964 895216052774896 860031680686064 433145314645463
454758089196737 441632677595089 371550774555605]
[ 92342146417517 0 583753158693405 603543734459023 540662194593155 930723353156714 284657638092236
423271773771732 312992807700385 459897744576492]
[ 389942908629505 377380128554073 28216769442888 591127799854154 316247602980563 754467704761660 770956076084520
298320288767464 918872495227747 204576691495555]
[ 408347756505642 406372427344417 612717698316805 299964748619746 591348231657186 302371740460318 421300098808982
0 173670142342295 69361901129395]
[ 324703201436906 981215793203770 204232473287550 496942343803851 355968231201706 865069609921374 1004588767307362
609138334283912 47116856113832 0]
[ 400619138252874 0 186640287370110 261566054772075 763646648268041 189457938907919 631286476979333
860377963801277 871676830062988 856013244432792]
[1030647789522790 679626823667083 0 381032100570714 451005439602351 400828544750347 977314407882773
860382258752061 97469403671661 851753710617944]]with h5py.File("<SN>.barcodeToPos.h5", "r") as f:
bp_matrix = f["bpMatrix_1"]
x, y = 6518, 12274
cid = int(bp_matrix[y, x, 0])
print("CID (decimal):", cid)Output:
CID (decimal): 1009044103032663.4.2 Convert CID to ATGC
As we mentioned in in blog of From CID to ATGC: Decoding Stereo-seq Barcodes, we can use the STOmics/ST_BarcodeMap to convert the CID numbers into the actual ATGC sequences, as shown below:
./ST_BarcodeMap-0.0.1 --in A02598A4.barcodeToPos.h5 --out barcodeToPos.txt --action 3Output:
ubuntu@ubuntu:~$ head barcodeToPos.txt
TGCTCTATCGCAACCCATGCTCCAG 26457 26459
GGATTACCACGTGTCCATTTACCCC 26456 26459
AATTGAAGCCGACACTGTATGGGGG 26453 26459
GCATATCGACCTCCTTCGATCCCTG 26447 26459
GTCGATGTGCTTGCAATCATGAGCT 26445 26459
AAGGTTTGCTTGAGCACGGGGCCGA 26444 26459
GAGCCTATGAACTTTCGCTCCGAAA 26443 26459
AAACTTAACAGCCGCCTGTCCTTTC 26442 26459
GCTCTCGGCGTCATCCATTTACTAC 26441 26459
GTCGTTTGTCCACGATAGCAACATT 26437 26459Then, we can use the file of tissue_xy_coords.txt obtained from Section 1 to get the barcodes inside the tissue:
awk 'NR==FNR {xy[$1"\t"$2]=1; next} ($2"\t"$3) in xy' tissue_xy_coords.txt barcodeToPos.txt > barcodeToPos_tissue.txtOutput:
ubuntu@ubuntu:~$ head barcodeToPos_tissue.txt
TTTCTGCCCCTTATAGCTGTTATCG 6436 26451
ACAAACCAACCTGTCTGTCCTGCGA 18777 26448
GACTATAACGGTAGCTTAGGGTCGT 14541 26448
CTTCATCGCTCGGTCCTCGCTTCTG 6066 26448
GCTTTCCCCAGCATCTCACGCACCT 14401 26446
CAACAGCCTTCCACACCTACGGCGA 5713 26441
CAGTGTCCTGAAAAATCGTTCTCTC 6599 26439
GCCCGCAAGCTCTCGCAAGCCTCAC 6194 26437
CGAGCGATATTGGCTCCGAGAAATT 19123 26435
GCATATCTAGGATAGCACTTAATTT 18867 26435Now, we can create a white list, barcodes_in_tissue.txt, of barcodes in tissue for later use:
cut -f1 barcodeToPos_tissue.txt > barcodes_in_tissue.txt3.4.3 Filter Read 1 FASTQ files
According to SAW User Manual V8.1, paired FASTQs include a pair of read files, read 1 for CID, MID information and read 2 for captured RNA sequencing data respectively. An example of paired FASTQ:
# read 1
@E100026571L1C001R00300000000/1
TGTCCAACGGAGACGGCTCCGACAAGGCACTGGCA
+
>DG;<BGH=>*EFE8*G/3E@2:F0-GBGG188F<
# read 2
@E100026571L1C001R00300000000/2
GTCTCACCATACTTTTACAAAGTTATTTCAACCCAAATCACAATTTAAGAATTATTTGTTCTACCTATGCCACACTTTAAATAAATGTCTATTAAAACCA
+
-GFEECG?ECBFF<=@A@<E@><;FGCF=>=E53FEF5>FGF@,0ADE9CEAG2GBE@HF3EA<CE;G2F@=G8=?@G9FBGE.EG6G2;974E*D9DE9Therefore, we can retrieve the all barcode-related reads ID from read 1 FASTQ file by using the extract_R1_ReadIDs_parallel.sh below:
#!/bin/bash
# Prerequisite: GNU parallel must be installed
# sudo apt install parallel
# === CONFIG ===
BARCODE_LIST="barcodes_in_tissue.txt"
OUTFILE="matched_read_ids.txt"
TMPDIR="tmp_matched_ids_parallel"
mkdir -p "$TMPDIR"
export BARCODE_LIST
export TMPDIR
# === FUNCTION TO PROCESS ONE FILE ===
process_file() {
fq="$1"
base=$(basename "$fq" _1.fq.gz)
tmpfile="$TMPDIR/${base}_ids.txt"
zcat "$fq" | paste - - - - \
| awk -v BARCODE="$BARCODE_LIST" -v TMP="$tmpfile" '
BEGIN {
while ((getline line < BARCODE) > 0) {
barcodes[line] = 1
}
}
{
seq = substr($2, 1, 25)
if (seq in barcodes) {
split($1, parts, "[/]")
readid = substr(parts[1], 2)
print readid >> TMP
}
}
'
}
export -f process_file
# === RUN IN PARALLEL ===
echo "🧠 Running parallel processing..."
ls *_1.fq.gz | parallel --bar -j $(nproc) process_file {}
# === MERGE & CLEANUP ===
echo "🧹 Merging and deduplicating..."
cat $TMPDIR/*_ids.txt | sort | uniq > "$OUTFILE"
rm -r "$TMPDIR"
echo "✅ Done. All matched read IDs saved to $OUTFILE"Output:
ubuntu@ubuntu:~$ head matched_read_ids.txt
E150018299L1C001R00100000800
E150018299L1C001R00100000806
E150018299L1C001R00100000892
E150018299L1C001R00100000906
E150018299L1C001R00100000969
E150018299L1C001R00100001174
E150018299L1C001R00100001181
E150018299L1C001R00100001294
E150018299L1C001R00100001305
E150018299L1C001R001000013113.4.4 Filter Read 2 FASTQ files
By using the matched_read_ids.txt from the above section, we can finally extract all reads inside the tissue by using extract_R2_parallel.sh below:
#!/bin/bash
# ====== CONFIG ======
IDFILE="matched_read_ids.txt"
OUTFILE="R2.tissue.fq.gz"
TMPDIR="tmp_r2_extract"
mkdir -p "$TMPDIR"
# ====== PREP ======
echo "📄 Indexing matched read IDs..."
sort "$IDFILE" | uniq > sorted_read_ids.txt
# ====== EXPORT FOR PARALLEL ======
export TMPDIR
export IDFILE="sorted_read_ids.txt"
# ====== FUNCTION TO PROCESS A SINGLE R2 FILE ======
extract_r2_reads() {
fq="$1"
base=$(basename "$fq" .fq.gz)
tmpout="$TMPDIR/${base}.tissue.fq"
zcat "$fq" | paste - - - - \
| awk -v IDFILE="$IDFILE" -v OUT="$tmpout" '
BEGIN {
while ((getline id < IDFILE) > 0) {
keep[id] = 1
}
}
{
split($1, a, "/");
readid = substr(a[1], 2); # remove leading @
if (readid in keep) {
print $1 "\n" $2 "\n" $3 "\n" $4 >> OUT
}
}
'
}
export -f extract_r2_reads
# ====== RUN PARALLEL ======
echo "🚀 Extracting from R2 FASTQs using parallel..."
ls *_2.fq.gz | parallel --bar -j $(nproc) extract_r2_reads {}
# ====== MERGE RESULTS ======
echo "🧬 Merging results into $OUTFILE..."
cat $TMPDIR/*.tissue.fq | gzip > "$OUTFILE"
# ====== CLEANUP ======
rm -r "$TMPDIR"
rm sorted_read_ids.txt
echo "✅ Done! Output written to $OUTFILE"
Output:
ubuntu@ubuntu:~$ zcat R2.tissue.fq.gz|head -n 12
@E150018299L1C001R00100000906/2
GTCTTAGGAAGACAATGTGAAGTTGGAGGTCGAGAGACCTCTGATAAGGTCGCCATGCCTCTCAGTACGTCAGCAGAAGCAGTGGTATCAACGCAGAGTA
+
DDDDDCCDDDDDDDDDCDDDDDDDDDCDCDDDCDCDCDDDDDDCDDCCCCDDDDCDDDDDCDDCDDDDDDDCCDCDCCDDCDCDDDCDDCCDDDCDCDDB
@E150018299L1C001R00100001311/2
CCTGATCAGTCTTAGGAAGACAAAGAAAAGTGGTGCCTAAGGCCTCACCTGATAAGGTCGCCATGCCTCTCAGTACGTCAGCAGAAGCAGTGGTATCAAC
+
BCDDCDCCCDCDDDCBAACCCDC@CCCCBDCDCCDCCDCCCBCCDBDBCDCCCCCDCCCDBBCCDCDDCCB>CDCBCCDCBCCC=<CCCCCBBACCC?BC
@E150018299L1C001R00100002321/2
ACAAAGCACCGAGGAAAAAAGTAATGAGTCTGATAAGGTCGCCATGCCTCTCAGTACGTCAGCAGAAGCAGTGGTATCAACGCAGAGTACATTCCGTTGA
+
CDDDCCDCCCDDDDDCCDCDDCCCDCCDCCDDCDCCDDDDDCCDDDCCDDDCDDDCDDDDCDDCDCCDDCCDDDDDDDCDDDDCDCDCCCDDCDCCDCDDReference:
🧾 TL;DR — Extract Tissue Reads from Stereo-seq Using GEF & XY Coordinates
This workflow extracts read sequences localized within tissue areas from Stereo-seq datasets. It starts by decoding spatial expression data in .tissue.gef, determines valid XY coordinates of tissue-covered areas, and then filters clean or raw FASTQ reads accordingly. The pipeline supports both pre-cleaned reads (--clean-reads-fastq) and unprocessed raw reads via barcode decoding from chip mask HDF5 files.
📄 Required Files
| 📁 File | 🧾 Description |
|---|---|
<SN>.tissue.gef | Gene expression matrix with spatial bins |
<SN>.barcodeToPos.h5 | Encoded CID matrix for raw coordinate mapping |
*_1.clean_reads.fq | Read1 files containing CID+MID+coordinates |
*_1.fq.gz, *_2.fq.gz | Raw paired FASTQs (Read1 for CID, Read2 RNA) |
🛠 Required Scripts
| ⚙️ Script | 🔧 Purpose |
|---|---|
explore_gef() | Inspects HDF5 GEF structure to locate XY coordinates |
filter_cleanR1_by_XY_parallel.sh | Filters clean Read1 FASTQs by tissue XY coords |
extract_R1_ReadIDs_parallel.sh | Gets read IDs from raw Read1 matching tissue barcodes |
extract_R2_parallel.sh | Extracts raw Read2 reads based on matched read IDs |
ST_BarcodeMap | Converts CID numbers to ATGC barcodes |
🧭 Workflow Summary
- Parse
.tissue.gefto extract XY expression data (geneExp/bin1/expression). - Save tissue XYs to
tissue_xy_coords.txt. - Use
--clean-reads-fastqRead1 files to filter by XY, outputtingR2.tissue.fq.gz. - For raw reads: decode barcodes using
barcodeToPos.h5+ST_BarcodeMap. - Match decoded barcodes to XY, extract read IDs, then filter raw Read2 FASTQs.
- Final output: clean or raw
R2.tissue.fq.gzwith sequences inside tissue.
📂 Output Files
| 📤 Output | 📌 Content |
|---|---|
tissue_xy_coords.txt | List of XY coords within tissue |
R2.tissue.fq.gz | Filtered FASTQ reads from tissue regions |
barcodes_in_tissue.txt | Whitelist of barcodes localized in tissue |
matched_read_ids.txt | Read IDs from Read1 sequences matching tissue barcodes |
flowchart TD
subgraph Input_Files
A1["📁 <SN>.tissue.gef"]
A2["📁 <SN>.barcodeToPos.h5"]
A3["📁 *_1.clean_reads.fq"]
A4["📁 *_1.fq.gz"]
A5["📁 *_2.fq.gz"]
end
subgraph Processing_Steps
B1["⚙️ explore_gef()"]
B2["📄 tissue_xy_coords.txt"]
B3["⚙️ ST_BarcodeMap"]
B4["📄 barcodeToPos.txt"]
B5["📄 barcodeToPos_tissue.txt"]
B6["📄 barcodes_in_tissue.txt"]
B7["⚙️ filter_cleanR1_by_XY_parallel.sh"]
B8["⚙️ extract_R1_ReadIDs_parallel.sh"]
B9["📄 matched_read_ids.txt"]
B10["⚙️ extract_R2_parallel.sh"]
end
subgraph Outputs
C1["📤 R2.tissue.fq.gz"]
end
A1 --> B1 --> B2
A2 --> B3 --> B4 --> B5 --> B6
B2 --> B7 --> C1
A3 --> B7
A4 --> B8 --> B9
B6 --> B8
A5 --> B10
B9 --> B10 --> C1
If you found this helpful, feel free to comment, share, and follow for more. Your support encourages us to keep creating quality content.