I made a silly story this week…
1. Background
I wanted to extract only the reads located within the tissue to reduce the FASTQ file size for running CARLIN analysis. These days, I rely heavily on AI. And this time, AI seriously misled me and wasted a lot of my time. Here’s what happened.
2. AI-explanations
The A02598A4.barcodeToPos.h5 file does not contain actual ATGC sequences, and I couldn’t find any documentation online about how to convert CID numbers into actual DNA barcodes. So, I turned to AI for help. AI responded:
Your integer CIDs are a compact representation of the CID DNA barcode. To use them for read filtering, you must convert the integer to the DNA sequence (typically 25 bases for Stereo-seq Bin50/100).
How the Encoding Works
- The integer is a base-4 encoding of the 25bp DNA CID barcode.
- A=0, C=1, G=2, T=3 (least significant base is at the end).
- You can convert any integer CID to its corresponding DNA sequence using a small script.
It’s pretty great, right? The AI also kindly provides me a python script for converting the CID numbers into the actual sequence:
1
2
3
4
5
6
7
8
9
10
11
```python
def cid_to_seq(cid_int):
base4 = []
for _ in range(25):
base = cid_int % 4
base4.append("ACGT"[base])
cid_int //= 4
return ''.join(reversed(base4))
print("25-mer:", cid_to_seq(cid_value))
```
In addition, AI backed its explanation with what seemed like credible references, as shown below:

I was convinced by the AI’s explanation after briefly skimming the online resources it cited, including:
- https://db.cngb.org/stomics/assets/html/stereo.seq.html
- Chen A, Liao S, Cheng M, Ma K, Wu L, Lai Y, Qiu X, Yang J, Xu J, Hao S, Wang X. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. 2022 May 12;185(10):1777-92.
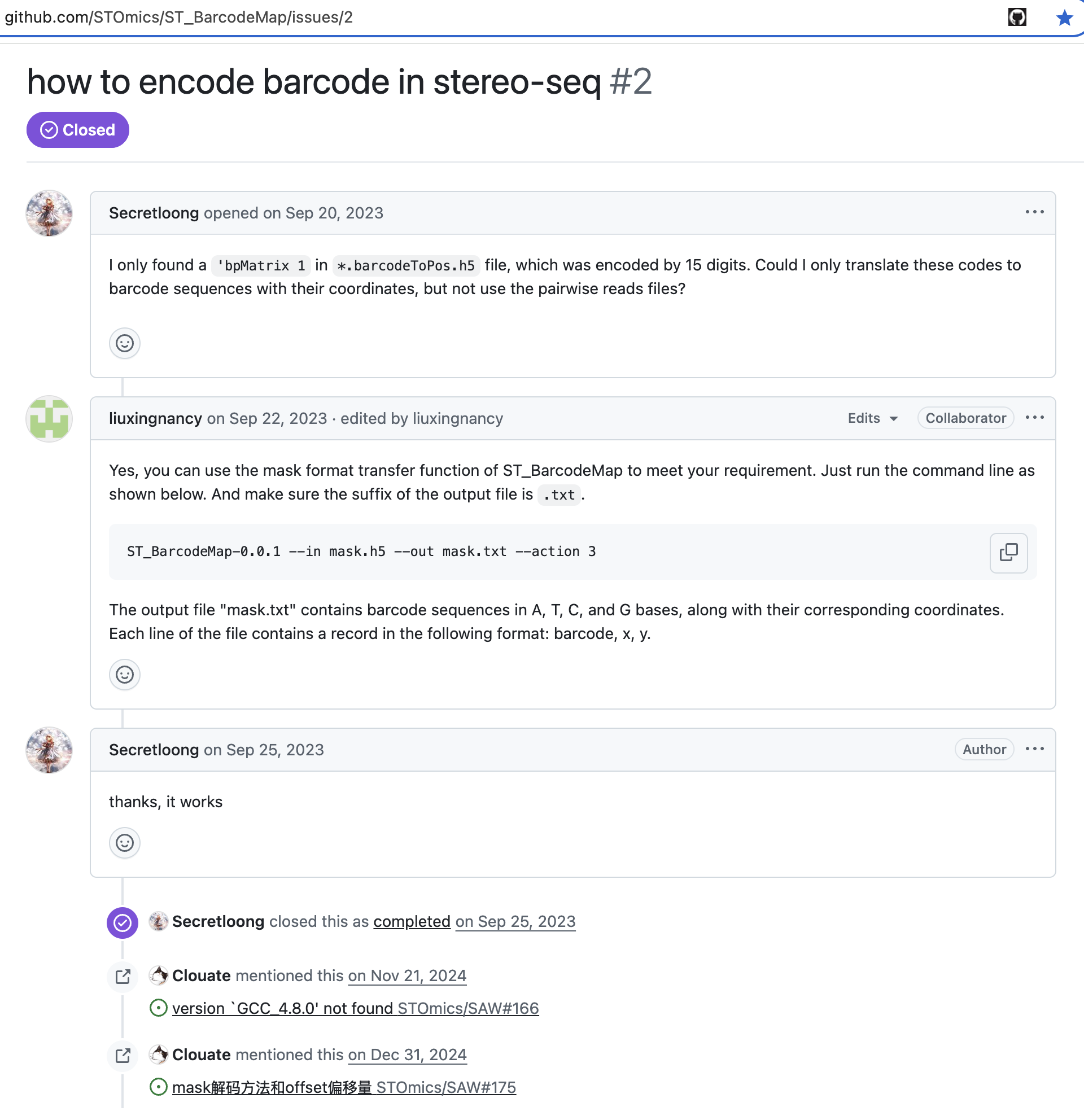
3. Verify AI’s CID2ATGC convert algorithm.
Fortunately, I’m not completely gullible—I had a moment of caution. I decided to validate AI’s algorithm using a read from the BAM file generated by MGI’s saw software. Let’s examine the example below:
- The below is a read record from bam file. I extract it by using the IGV software:
Read name = E150018299L1C036R00400117279
Read length = 100bp
Flags = 0
----------------------
Mapping = Primary @ MAPQ 255
Reference span = chr3:129,504,132-129,504,231 (+) = 100bp
Cigar = 100M
Clipping = None
----------------------
XF = 4
NH = 1
HI = 1
nM = 0
UR = 6B507
AS = 98
Cx = 6518
Cy = 12274
---
Location = chr3:129,504,138
Base = A @ QV 34
- A bash script below,
./search.sh, was used to search the read name of “” across all 16 R1.fq.gz files of my stereo-seq:
1
2
3
4
5
```bash
for f in *_1.fq.gz; do
zgrep -A1 'E150018299L1C036R00400117279' "$f" && echo "✅ Found in $f" && break
done
```
- The above bash script returns:
🔎 Searching E150018299_L01_62_1.fq.gz [ 6/ 16]
✅ Found in file: E150018299_L01_62_1.fq.gz @E150018299L1C036R00400117279/1 TATAGTACATATGATTCCAGGTCCACGGTCCAACT
🔍 Search complete.
- The AI’s algorithm returns:
1
2
3
4
5
6
7
8
9
```python
import h5py
import numpy as np
with h5py.File("./A02598A4/00.Rawdata/mask/A02598A4.barcodeToPos.h5", "r") as f:
bp_matrix = f["bpMatrix_1"]
x, y = 6518, 12274
cid = int(bp_matrix[y, x, 0])
print("CID (decimal):", cid)
```
CID (decimal): 100904410303266
1
2
3
4
5
6
7
8
9
10
11
```python
def cid_to_seq(cid_int):
base4 = []
for _ in range(25):
base = cid_int % 4
base4.append("ACGT"[base])
cid_int //= 4
return ''.join(reversed(base4))
print("25-mer ACGT:", cid_to_seq(cid))
```
25-mer ACGT: ACCGTTACCGGATGAGACAGTAGAG
Actual sequence (TATAGTACATATGATTCCAGGTCCACGGTCCAACT) versus AI’s algorithm (ACCGTTACCGGATGAGACAGTAGAG)…
There is no match between the AI’s algorithm-derived 25-mer (ACCGTTACCGGATGAGACAGTAGAG) and the actual R1 sequence (TATAGTACATATGATTCCAGGTCCACGGTCCAACT).
4. The More Artificial, the More Intelligent
Feeling disheartened, I decided to give Google one more careful search—and that’s when I saw it:
Well… that’s life, isn’t it?
5. Not end yet…
Do you think it is over? No! My Linux environment do not meet the compile prerequisites of ST_BarcodeMap below:
- linux
- gcc-8.2.0
- boost >=1.73.0
- zlib >=1.2.11
- hdf5 >=1.10.7
And after testing, we need boost==1.73.0 not >1.73.0
Then, I used conda with mamba to build an environment to meet there prerequisites:
🟢
mambais a drop-in replacement forconda, but drastically faster for solving.
conda install mamba -n base -c conda-forge
mamba create -n barcodeenv boost=1.73 hdf5=1.10.7 zlib=1.2.11 -c conda-forge
conda activate barcodeenv
mamba install -c conda-forge gcc_linux-64 gxx_linux-64
mamba install -c conda-forge boost=1.73
However, the makefile of ST_BarcodeMap does not support to use conda environment. So I have to modify its makefile as below:
1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
```bash
DIR_INC := ./inc
DIR_SRC := ./src
DIR_OBJ := ./obj
PREFIX ?= /usr/local
BINDIR ?= $(PREFIX)/bin
# Use Conda’s Boost, but system GCC
INCLUDE_DIRS = -I$(DIR_INC) -I$(CONDA_PREFIX)/include -I/usr/include/hdf5/serial
LIBRARY_DIRS = -L$(CONDA_PREFIX)/lib -L/usr/lib/x86_64-linux-gnu/hdf5/serial
SRC := $(wildcard ${DIR_SRC}/*.cpp)
OBJ := $(patsubst %.cpp,${DIR_OBJ}/%.o,$(notdir ${SRC}))
TARGET := ST_BarcodeMap-0.0.1
BIN_TARGET := ${TARGET}
CXX := g++
CXXFLAGS := -std=c++11 -g -O3 $(INCLUDE_DIRS)
LD_FLAGS := $(LIBRARY_DIRS) -lboost_serialization -lhdf5 -lz -lpthread
${BIN_TARGET}: ${OBJ}
$(CXX) $(OBJ) -o $@ $(LD_FLAGS)
${DIR_OBJ}/%.o: ${DIR_SRC}/%.cpp make_obj_dir
$(CXX) -c $< -o $@ $(CXXFLAGS)
.PHONY: clean
clean:
rm -f ${DIR_OBJ}/*.o
rm -f ${TARGET}
make_obj_dir:
@if [ ! -d ${DIR_OBJ} ]; then mkdir -p ${DIR_OBJ}; fi
install:
install ${TARGET} ${BINDIR}/${TARGET}
@echo "Installed."
```
Now, I can happily compile the ST_BarcodeMap finally:
export LD_LIBRARY_PATH=$CONDA_PREFIX/lib:$LD_LIBRARY_PATH
cd ST_BarcodeMap
make
Finally, I got the actual ATGC sequence barcode for CID numbers.
./ST_BarcodeMap-0.0.1 --in A02598A4.barcodeToPos.h5 --out barcodes.txt --action 3
For the complete code and workflow related to my original goal, check out the entry
👉 Extract Tissue CIDs from Stereo-seq
in our BSGOU public wet/dry-lab notebooks online.
However, using the GitHub Actions workflow to compile the project is actually much easier and faster than setting it up and compiling it locally. Therefore, I forked the original ST_BarcodeMap repository and compiled it directly through GitHub.
You can check out my forked repository here: wong-ziyi/ST_BarcodeMap.
In the end, bioinformatics is still part biology, part informatics… and part “don’t blindly trust the robot” as Apple just found no evidence of formal reasoning in language models.
References
- https://db.cngb.org/stomics/assets/html/stereo.seq.html
- Chen A, Liao S, Cheng M, Ma K, Wu L, Lai Y, Qiu X, Yang J, Xu J, Hao S, Wang X. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell. 2022 May 12;185(10):1777-92.
- https://github.com/STOmics/ST_BarcodeMap/issues/2
- https://github.com/STOmics/ST_BarcodeMap
- The Illusion of Thinking: Understanding the Strengths and Limitations of Reasoning Models via the Lens of Problem Complexity
If you found this helpful, feel free to comment, share, and follow for more. Your support encourages us to keep creating quality content.